

在重组蛋白生产过程中,生物污染一直是一个不容忽视的问题。生物污染的来源多种多样,细菌、真菌、支原体、病毒等生物污染会造成严重的药品质量事件,威胁用药患者生命安全。因此,对已生产的药品或正在生产的药品,我们要尽量准确的检测其中的微生物与病毒的生物负荷,避免受到污染药品流入市场。
汉腾生物拥有1200+㎡的GMP质量控制实验室,拥有国际一流的先进设备,具备符合ChP, USP, EP等药典要求的检测能力,满足全面的数据可靠性要求。
汉腾生物重组蛋白生产平台拥有自主开发的高表达CHO细胞株构建平台,为客户提供专业的双抗特异性抗体(BsAb)和重组蛋白生产服务。

汉腾生物车间生产工艺流程图
整个生产过程中,原材料(生物来源),细胞库(主细胞库,工作细胞库),未加工原液物料(生物反应器收获前),下游工艺的适当阶段,原液等需要进行微生物限度检测,以确认各工序中微生物负荷在规定范围内。
药品生产过程中有关产品的微生物控制类型主要包括无菌检测、微生物限度检测和支原体检测。
无菌检测
无菌检查法系用于检查药典要求无菌的药品、生物制品、医疗器械、原料、辅料及其他品种是否无菌的一种方法。若供试品符合无菌检查法的规定,仅表明了供试品在该检验条件下未发现微生物污染。无菌产品的最终放行需遵循各国法规,《中国药典》2020版1101无菌检测法,USP-NF <71> STERILITY TESTS,EP 2.6.1,JP 4.06均对产品无菌检测进行了规定,其中《中国药典》2020版在无菌检查方面的要求,完善了灭菌法和无菌检查用隔离系统的验证及应用。
无菌检测的方法主要包括直接接种法和薄膜过滤法两种:

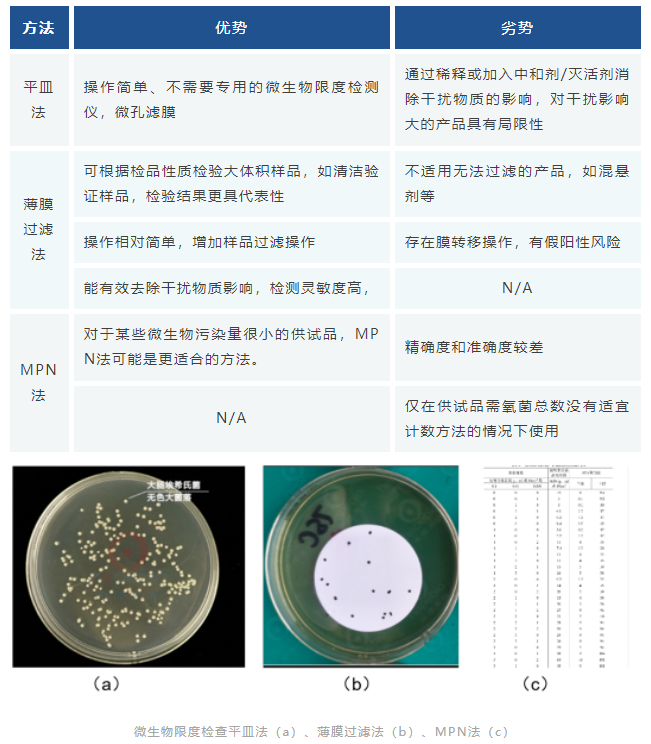
微生物限度检查
本法适用非无菌产品的微生物限度检测,检验全过程必须严格遵守无菌操作,防止再污染。防止污染的措施不得影响供试品中微生物的检出。洁净空气区域、工作台面及环境应定期进行监测。如供试品有抗菌特性,应尽可能去除或中和。
微生物限度的检测,《中国药典》2020版1105&1106&1107,USP-NF <61> &<1111>,EP 2.6.12&5.1.4等对非无菌产品微生物限度检查进行了详细规定,其中《中国药典》2020版明确了半固体制剂的微生物限度标准,半固体制剂与液体制剂一样应从严控制。
非无菌产品微生物限度检测的方法主要包括平皿法、薄膜过滤法和MPN法三种:
支原体检测
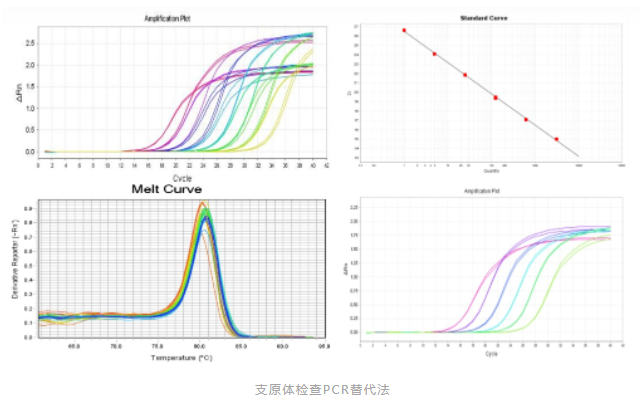
2020版《中国药典》3301 支原体检查法中规定,在进行主细胞库、工作细胞库、病毒种子批、对照细胞以及临床治疗用细胞需进行支原体检查时,应同时进行培养法和指示细胞培养法(DNA染色法)。病毒类疫苗的病毒收获液、原液采用培养法检查支原体;必要时,亦可釆用指示细胞培养法筛选培养基。也可采用经国家药品检定机构认可的其他方法。在进行支原体检验时,需要建立专门的支原体检验室,以防止样品的交叉污染。
采用传统的方法进行支原体检查,其检测周期长,培养法至少4周,指示细胞法至少2周,对于某些药物,病人亟需使用来挽救生命时,传统方法不再适用。2020版《中国药典》9201 药品微生物检验替代方法验证指导原则,USP-NF 〈1071〉 RAPID MICROBIAL TESTS FOR RELEASE OF STERILE SHORT-LIFE PRODUCTS: A RISK-BASED APPROACH(无菌短货架期产品放行的快速微生物检查法)以及EP 2.6.7(2007年首次提出用PCR技术进行支原体检测的指导纲要)均提出替代检验方法的概念,即“在控制药品微生物质量中,微生物实验室出于各种原因,如成本、生产量、快速简便及提高药品质量等需要而采用非药典规定的检验方法(即替代方法)时,应进行替代方法的验证,确认其应用效果优于或等同于药典的方法。”
微生物污染溯源调查
无菌检测、微生物检测及支原体检测都是培养的方法来确定样品是否被微生物污染。培养法更多依赖于人员的熟练操作,操作人员经验不足或操作不规范易形成假阳性,样品检验方法的不适用,又易形成假阴性的结果。假阴性与假阳性的结果严重影响检测结果的准确性,为患者用药安全带来极大隐患。
对于假阴性结果,若不能及时发现,则会导致污染批次产品正常放行,危及病人安全,最终导致产品召回,品牌受损,甚至工厂停工或倒闭。为避免假阴性的结果,更多的是对检验过程规范性,人员上岗操作能力的要求。
对于假阳性结果,如果没有找到造成污染的根本原因,法规是禁止进行重复测试的,这就需要大量的调查以找到根源,若无法调查出根本原因,最终可能导致该批次产品的报废。这就要求我们对微生物阳性检测结果进行污染调查。
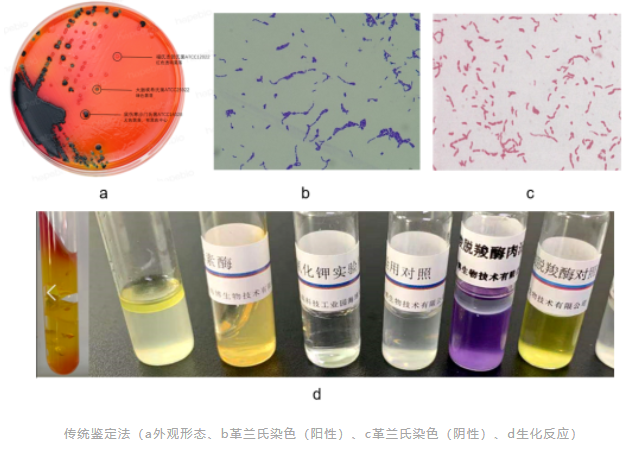
《中国药典》2020版 9204 微生物鉴定指导原则中指出,“大多数非无菌药品生产过程和部分无菌生产环境的风险评估中,对所检出微生物的常规特征包括菌落形态学、细胞形态学(杆状、球状、细胞群、孢子形成模式等)、革兰染色或其他染色特性,及某些能够给出鉴定结论的关键生化反应(如氧化酶、过氧化氢酶和凝固酶反应)进行分析,一般即可满足需要”、“非无菌产品的控制菌检查一般应达到药典规定的水平”、“无菌试验结果阳性、无菌生产模拟工艺(如培养基灌装)失败、环境严重异常事件时,对检出的微生物鉴定至少达到种水平,必要时需达到菌株水平”。
传统的表型鉴定技术包括观察菌落的形态(如大小、颜色等)、染色、显微镜检查以及生化鉴定等,微生物的表型具有一定的可变性,此外,传统的表型鉴定技术依赖人员经验和培养条件,其在一些难以培养、生长缓慢或需要特殊营养的微生物鉴定方面的局限性大。
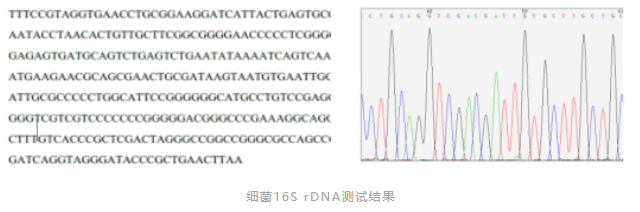
在应对药品微生物污染事件时,传统方法已不能满足快速分型、准确鉴定、溯源分析的需要。因此,以分子生物学为代表的基因型鉴定方法逐渐得到应用。《中国药典》2020版,对无菌或微生物检测阳性异常结果的调查,新增了1021细菌DNA特征序列鉴定法。如细菌16SrDNA测序技术、真菌18SrDNA测序技术。
微生物的溯源分析中涉及的病原微生物操作及鉴定,应该生物安全二级实验室中进行(P2),汉腾生物已于2022年取得P2实验室资质,可承接相关病原微生物的分离、纯化、保种及鉴定等工作。
2017年中国加入ICH,中国医药行业充分利用中国加入ICH全面与国际接轨的时机,进一步借鉴国内外成熟的、先进的检测技术和经验、结合我国药品生产和质量控制现状,加强通用性技术要求的制定以及国际协调和统一。
汉腾生物QC拥有先进的自动化检验设备,配备了足够的专业检测人员,具有重组蛋白、抗体、疫苗等方面的检测能力以及相关经验,建立了完善的生产前、生产中及生产后产品质量控制策略。具备的检测能力包括:
HPLC/UPLC类:SEC, CEX, 肽图,PR-HPLC;
电泳类:CE-SDS, cIEF, SDS-PAGE, 核酸琼脂糖电泳,IEF
免疫学类:ELISA活性,免疫印迹,免疫电泳,HCP(CHO,E.coli, 人源细胞),SelectVII 残留
生物学活性类:细胞增殖抑制(MTT),流式细胞法
微生物类:微生物限度,无菌,细菌内毒素(凝胶法,动态法)
其他常规理化类:不溶性微粒,TOC,渗透压,密度,蛋白含量(UV, Brandford),水分,化学滴定反应,薄层色谱……
具备BLS-2实验室资质,符合生物安全的法规与要求。